APÉNDICE SOBRE FISIOLOGÍA CELULAR
Bioenergética y metabolismo, parte I: Termodinámica desde la biología celular
La termodinámica es una de esas ramas sumamente mal interpretadas fuera de su campo de aplicación físico, lo cual fácilmente se podría decir de cualquier otra subdisciplina científica, de no ser porque la termodinámica es una de las más abusadas para toda clase de metáforas y especulaciones acerca de la consciencia, la conducta, la vida, la información y hasta la apologética cristiana; unas peores que otras. El propósito de esta entrada no es explicar lo que está mal con la termodinámica como la emplean los biólogos celulares, sino simplemente explicitar la manera en que se enseña y se utiliza, empleando como caso de estudio dos libros de texto introductorios (Alberts et al., 2014; Cooper, 2019) y un artículo científico.
La energía total de un sistema y su entorno permanece constante (Cooper 2019), lo cual normalmente se traduce a la energía no se crea ni se destruye, tan solo se transforma: se convierte de un tipo a otro. Piénsese en los intercambios entre energía cinética y potencial al estudiar la trayectoria de una partícula en descenso vertical. A medida que la partícula cae, su energía gravitacional potencial se reduce mientras que su energía cinética aumenta. En un ejemplo más biológico, la energía química es “convertida” en energía mecánica, como lo que ocurre en la contracción de un músculo.
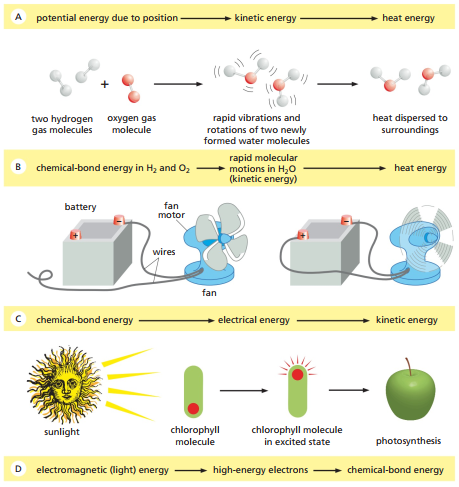
La Figura 1 ilustra diferentes formas de cambios en tipos de energía, usando primero el ejemplo de un ladrillo en picada. A medida que el ladrillo está suspendido sobre la superficie, este posee una energía potencial gravitacional que se va convirtiendo en energía cinética a medida que desciende, hasta que, al caer, ‘transforma’ su energía cinética en calor liberado.
Del mismo modo, la Figura 1 ilustra un caso donde energía química es almacenada en moléculas de gas e hidrógeno. La cantidad de energía liberada cuando se forman las moléculas de agua lleva a que estas vibren y, por medio de colisiones espontáneas entre moléculas de agua, la energía es eventualmente transferida como calor al entorno.
Figura 1. Las células vivas no ‘desafían’ la segunda ley de la termodinámica. En la izquierda, las moléculas tanto dentro como fuera de la célula exhiben un grado de desorden aparente. La célula, a través del metabolismo, ordena las moléculas en su interior mientras que aumenta la cantidad de desorden en el entorno a través de la liberación de calor (Alberts et al., 2014, p. 87).
El grado de desorden (o entropía) en un sistema incrementa con el paso del tiempo (Cooper, 2019). Cooper no entra en detalle sobre lo que el desorden o la entropía implica, tan solo esperando que sea comprensible a través de una serie de ejemplos biológicos. Intuitivamente, afirma Cooper, las células parecen violar la segunda ley en la medida que los precursores de sus componentes son menos ordenados: las proteínas son polímeros ordenados de aminoácidos, por lo cual la síntesis de proteína implica una reducción (en vez de un aumento) de entropía.
Más allá de la definición de desorden o entropía implicada en el texto, Cooper resuelve el fallo intuitivo haciendo referencia a que las células son sistemas abiertos, por lo cual intercambian energía con el entorno. Esto quiere decir que el aumento de entropía puede ser balanceado a través del entorno. La totalidad sistema-entorno puede aumentar su entropía sin que el incremento se vea estrictamente manifestado en el sistema celular en sí mismo. Cooper (2019) afirma que una reacción química reduce la entropía del sistema en tanto el calor liberado por la reacción sea suficiente para generar un incremento mayor de entropía en el entorno (el cual compense por el orden generado dentro de la célula). La Figura 2 ilustra cómo el aumento de ‘orden’ en la célula implica un aumento de ‘desorden’ en el entorno.
Figura 2. Las células vivas no ‘desafían’ la segunda ley de la termodinámica. En la izquierda, las moléculas tanto dentro como fuera de la célula exhiben un grado de desorden aparente. La célula, a través del metabolismo, ordena las moléculas en su interior mientras que aumenta la cantidad de desorden en el entorno a través de la liberación de calor (Alberts et al., 2014, p. 86).
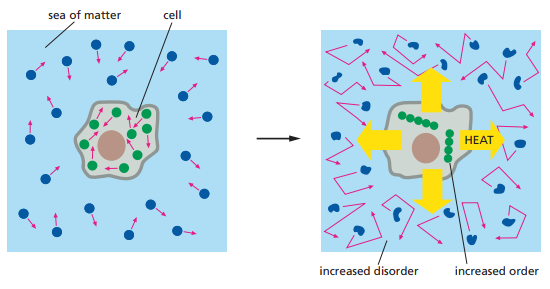
Aunque Cooper (2019) no define la entropía con claridad, Alberts et al. (2014) intentan dilucidarla a través de una apelación intuitiva a la probabilidad y a ejemplos pedagógicos paradigmáticos como el del cuarto desordenado que requiere de “energía” para ser arreglado por su dueño. La manera en que Alberts et al. (2014) construyen la entropía la hace ver como una medida escalar del desorden, mientras que el desorden es explicado en términos de la probabilidad del arreglo de los componentes de un sistema.
A modo de ejemplo, los autores pintan la imagen de una caja con 100 monedas en cara, y afirman que si la caja es sacudida, esta tenderá a un arreglo donde las monedas estarán ordenadas en una mezcla de 50 caras y 50 cruces, ya que este arreglo tiene mayor probabilidad que otros donde, digamos, todas sean caras otra vez, o todas sean cruz, o sean más de una que de otra. La conclusión que los autores construyen para sus lectores es que la entropía es una medida del desorden, y que a mayor el desorden mayor la entropía:
“We can express the second law in terms of probability by stating that systems will change spontaneously toward those arrangements that have the greatest probability. Consider a box of 100 coins all lying heads up. A series of events that disturbs the box will tend to move the arrangement toward a mixture of 50 heads and 50 tails. The reason is simple: there are a huge number of possible arrangements of the individual coins that can achieve the 50–50 result, but only one possible arrangement that keeps them all oriented heads up. Because the 50–50 mixture accommodates a greater number of possibilities and places fewer constraints on the orientation of each individual coin, we say that it is more “disordered.” [...] The measure of a system’s disorder is called the entropy of the system, and the greater the disorder, the greater the entropy" (Alberts et al., 2014, p. 85).
La energética de las reacciones bioquímicas es descrita en términos de la función deltada de la energía libre de Gibbs (G) (Cooper, 2019). La Ecuación 1 ilustra que el cambio en energía libre de Gibbs, o ΔG, es igual al cambio en entalpía (ΔH) menos el producto de la temperatura absoluta (T) por el cambio en entropía (ΔS).
La entalpía se refiere al calor liberado o absorbido durante una reacción química.
En el caso químico hipotético donde A pueda ser convertido a B:
Las reacciones químicas ocurrirán espontáneamente en la dirección energéticamente favorable, es decir, aquella que implique una reducción de energía libre (ΔG < 0). Entonces, si para A → B la reacción implica ΔG < 0, entonces la reacción ocurrirá en la dirección hacia delante, mientras que si la reacción implica ΔG > 0 esta irá en dirección contraria. Es decir, se favorecen direcciones de reacciones químicas donde ΔG es negativo. Esto se debe a que la segunda ley de la termodinámica necesita que las reacciones químicas procedan solo si el resultado implica un aumento en el ‘desorden’ del universo. Por ello, solo cuando energía libre de Gibbs es perdida se favorece esa dirección espontáneamente. Así, las reacciones energéticamente favorables son aquellas que aumentan el desorden a través de la reducción energía libre de Gibbs.
Puesto que el ΔG de una reacción es una función tanto de las propiedades intrínsecas de los reactivos y los productos como de las condiciones del entorno (temperatura, concentraciones), los cambios de energía libre se definen bajo condiciones estándares: concentración 1-M de todos los reactivos y productos y 1 atm de presión.
En condiciones estándares, se puede determinar el cambio estándar de energía libre de Gibbs (ΔG°) de una reacción. Una vez definido el estado estándar, ΔG° se entiende convencionalmente como una propiedad característica de la reacción química misma.
Ahora bien, con un entendimiento de ΔG° y la razón de concentración [B]/[A] para una reacción A→B dada, es posible también determinar ΔG ya que este es una función de ambas. Así, la Ecuación 2 ilustra un segundo método para determinar ΔG si se conoce ΔG° para la reacción:
En el caso químico hipotético donde A pueda ser convertido a B:
Es decir, el cambio de energía libre de Gibbs es igual a la sumatoria de dos términos, el primero siendo el cambio estándar de energía libre de Gibbs de la misma reacción química más el producto de la constante de gas (R) por la temperatura absoluta (T) y el logaritmo natural de la razón de las concentraciones de B y A.
En el caso donde ΔG sea igual a 0, esto quiere decir que la reacción no procede con una preferencia direccional, lo cual se conoce como punto de equilibrio. Es decir, la reacción ocurre en tasas iguales en ambas direcciones. Para que ΔG sea igual a 0, ΔG° y RT ln [B]/[A] deben ser iguales en magnitud y contrarios en signo. Defínase la concentración a la que ΔG=0 como K=[B]/[A], así, adquirimos la Ecuación 3:
En el caso químico hipotético donde A pueda ser convertido a B:
Lo cual nos arroja la Ecuación 4:
En el caso químico hipotético donde A pueda ser convertido a B:
Y en este sentido RT ln K está directamente relacionado a ΔG°, siendo igual en magnitud y opuesto en signo.
Así, si la razón real [B]/[A] es mayor que la razón de equilibrio (K), ΔG > 0, por lo cual la reacción ocurrirá en la dirección contraria (de B hacia A). Por último, si [B]/[A] es menor que K, entonces ΔG < 0, lo cual implica que se favorecerá la reacción de A hacia B.
Para propósitos bioquímicos, ΔG° no es particularmente útil ya que las condiciones celulares difieren. Por ello, se emplea el cambio en energía libre de Gibbs ΔG°’, el cual implica una solución acuosa de pH = 7.
Bajo condiciones celulares, numerosas reacciones biológicas son termodinámicamente desfavorables (ΔG > 0). Por ello, para que estas reacciones ocurran debe existir una fuente de energía adicional. Dada la siguiente reacción:
La conversión de A a B es energéticamente desfavorable, por lo cual la reacción tiende netamente al lado contrario. No obstante, al acoplar esta reacción con la siguiente:
Ambas reacciones toman la siguiente forma:
La sumatoria de ΔG para ambas reacciones produce un ΔG neto menor a 0, lo cual hace que la primera reacción considerada se vuelva energéticamente favorable. En las células, las enzimas son las que se encargan de ejecutar estas reacciones acopladas energéticamente favorables para producir biomoléculas que, de otra manera, implican cambios energéticos desfavorables.
Ahora bien, para que las enzimas operen de esta manera, es necesaria una reacción química capaz de liberar suficiente energía libre de Gibbs para impulsar la ocurrencia de otras reacciones energéticamente desfavorables. En las células, este papel es tomado por el adenosín trifosfato (ATP).
Cooper (2019) afirma que el ATP sirve como un “almacén” de energía libre dentro de la célula (Figura 3). Químicamente hablando, no hay nada especial en los enlaces de fosfato. La razón por la que son llamados enlaces de alta energía radica en que, al ser hidrolizados dentro de la célula, liberan grandes cantidades de energía libre de Gibbs.
Figura 3. Los enlaces entre los grupos de fosfato del ATP se describen como enlaces de “alta energía” ya que la hidrólisis resulta en una reducción sustancial de energía libre de Gibbs. El ATP puede ser hidrolizado a ADP más un grupo fosfato (HPO42-) o AMP más un pirofosfato. A su vez, el pirofosfato puede ser hidrolizado para liberar más energía (Cooper, 2019, p. 84).
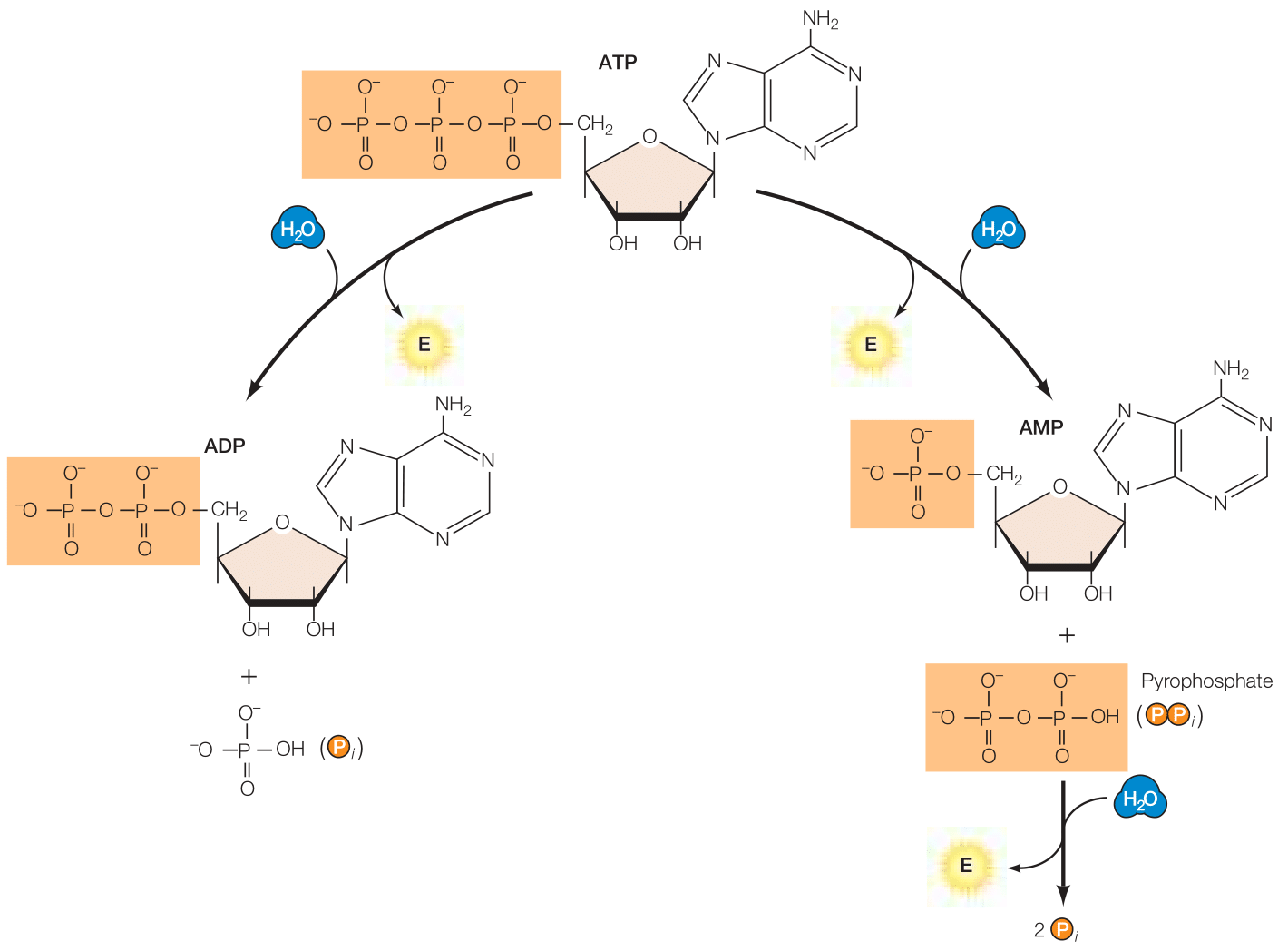
En la hidrólisis de ATP a adenosín difosfato (ADP) más fosfato (Pi), el cambio energético es de ΔG°’ = -7.3 kcal/mol. Ahora bien, las diferencias en concentración intracelular al comparar el cambio en energía libre en condiciones estándares con los de una célula en estado normal arroja que la cantidad de energía liberada es mucho mayor, de ΔG = -12 kcal/mol.
Así como el ATP puede ser hidrolizado a ADP + Pi, también puede ser hidrolizado a adenosín monofosfato (AMP) más pirofosfato (PPi). Esta reacción, según Cooper (2019), cede prácticamente la misma cantidad de energía libre de Gibbs que la hidrólisis de ATP a ADP + Pi, con la diferencia de que el pirofosfato en sí mismo puede ser rápidamente hidrolizado con un ΔG similar a la hidrólisis del ATP a ADP.
En función de esta reducción en la energía libre por la hidrólisis de ATP, esta reacción química sirve para impulsar otras reacciones químicamente desfavorables a través de un acoplamiento. A modo de ejemplo, el primer paso en la glucólisis, que forma una glucosa-6-fosfato (G6P) a partir de una glucosa y un grupo fosfato, es energéticamente desfavorable:
Para poder impulsar esta reacción química, debe ser acoplada con la hidrólisis de ATP:
La reacción combinada toma la siguiente forma:
Otros trifosfato de nucleósidos también poseen enlaces de alta energía, por lo cual pueden ser usados de la misma forma que el ATP. No obstante, para la gran mayoría de las reacciones energéticamente desfavorables el ATP facilita la energía libre necesaria para impulsarlas.
Además de los libros de texto, cabe resaltar cómo se emplea la termodinámica en el lenguaje y el tipo de posturas a las que se invita implícita e inconscientemente a través de este:
Por una parte, el enmarque de las células es teleonómico. El lenguaje pedagógico sugiere que las células operan bajo criterios de logro, lo cual implica normatividad funcional: existen estados correctos y otros fallidos para la célula. Si bien esto no implica necesariamente una forma de intención, este lenguaje permite y autoriza el desarrollo de posturas teleológicas sobre la actividad celular. Aunado al tratamiento de la célula como capaz de “regular” y “mantener” su propio “desorden”, el resultado es la construcción de la célula como un agente causal normativo.
El discurso agencial, causal y normativo de la célula se extiende en la medida que se afirma que las células tienen control de regulación y mantenimiento de su “orden y desorden”, tomando en cuenta que la base explicativa de estos términos son las probabilidades. Las probabilidades en la mecánica estadística no son propiedades inherentes de los sistemas, así como la medida de entropía como inaccesibilidad o falta de información de los gases no es una propiedad de estos, sino de lo que sabemos sobre ellos. A modo de ejemplo, reducir el volumen V de un gas ideal también reduce su entropía (en términos de información) porque reduce la cantidad de microestados posibles y, por ende, la falta de información que tenemos sobre el gas. Esto tiene como consecuencia que atribuir la capacidad de regular y mantener su propio orden implica que la célula actúa como si tuviera conocimiento sobre las probabilidades de su propio sistema, lenguaje que legitima la atribución de mecanismos de procesamiento de información y computacionales a esta.
Alberts, B., Bray, D., Hopkin, K., Johnson, A., Lewis, J., Raff, M., Roberts, K., & Walter, P. (2014). Essential cell biology (4th ed.). Garland Science
Cooper, G. M. (2019). The cell: A molecular approach (8th ed.). Oxford University Press
Freddy J. Molero-Ramírez
fmolero@mail.uniatlantico.edu.co